Lysosomal Stress in Cardiovascular Diseases: Therapeutic Potential of Cardiovascular Drugs and Future Directions.
Study Design
- Çalışma Türü
- Review
- Popülasyon
- None
- Müdahale
- Lysosomal Stress in Cardiovascular Diseases: Therapeutic Potential of Cardiovascular Drugs and Future Directions. None
- Karşılaştırıcı
- None
- Birincil Sonuç
- None
- Etki Yönü
- Mixed
- Yanlılık Riski
- Unclear
Abstract
Lysosomal dysfunction has emerged as a central contributor to the pathogenesis of cardiovascular diseases (CVDs), particularly due to its involvement in chronic inflammation, lipid dysregulation, and oxidative stress. This review highlights the multifaceted roles of lysosomes in CVD pathophysiology, focusing on key mechanisms such as NLRP3 inflammasome activation, TFEB-mediated autophagy regulation, ferroptosis, and the role of apolipoprotein M (ApoM) in preserving lysosomal integrity. Additionally, we discuss how impaired lysosomal acidification, mediated by V-ATPase, contributes to lipid-induced cardiac dysfunction. Therapeutically, several pharmacological agents, such as statins, SGLT2 inhibitors, TRPML1 agonists, resveratrol, curcumin, and ferroptosis modulators (e.g., GLS1 activators and icariin), have demonstrated promise in restoring lysosomal function, enhancing autophagic flux, and reducing inflammatory and oxidative injury in both experimental models and early clinical settings. However, key challenges remain, including limitations in drug delivery systems, the absence of lysosome-specific biomarkers, and insufficient clinical validation of these strategies. Future research should prioritize the development of reliable diagnostic tools for lysosomal dysfunction, the optimization of targeted drug delivery, and large-scale clinical trials to validate therapeutic efficacy. Incorporating lysosome-modulating approaches into standard cardiovascular care may offer a new precision medicine paradigm for managing CVD progression.
Kısaca
This review highlights the multifaceted roles of lysosomes in CVD pathophysiology, focusing on key mechanisms such as NLRP3 inflammasome activation, TFEB-mediated autophagy regulation, ferroptosis, and the role of apolipoprotein M (ApoM) in preserving lysosomal integrity.
Full Text
Lysosomal Stress in Cardiovascular Diseases: Therapeutic Potential of Cardiovascular Drugs and Future Directions
Toshiki Otoda 1,2,* , Ken-ichi Aihara 2 and Tadateru Takayama 1
- 1 Division of General Medicine, Department of Internal Medicine, Nihon University School of Medicine, 30-1 Oyaguchikamicho, Itabashi, Tokyo 173-8610, Japan; [email protected]
- 2 Department of Community Medicine and Medical Science, Tokushima University Graduate School of Biomedical Sciences, 3-18-15, Kuramoto-cho, Tokushima 770-8503, Japan; [email protected]
* Correspondence: [email protected]; Tel.: +81-3-3972-8111
Academic Editors: Celestino Sardu and Alfredo Caturano
Received: 18 January 2025 Revised: 29 March 2025 Accepted: 24 April 2025 Published: 27 April 2025
Citation: Otoda, T.; Aihara, K.-i.; Takayama, T. Lysosomal Stress in Cardiovascular Diseases: Therapeutic Potential of Cardiovascular Drugs and Future Directions. Biomedicines 2025, 13, 1053. https://doi.org/10.3390/ biomedicines13051053
Copyright: © 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/ licenses/by/4.0/).
Abstract: Lysosomal dysfunction has emerged as a central contributor to the pathogenesis of cardiovascular diseases (CVDs), particularly due to its involvement in chronic inflammation, lipid dysregulation, and oxidative stress. This review highlights the multifaceted roles of lysosomes in CVD pathophysiology, focusing on key mechanisms such as NLRP3 inflammasome activation, TFEB-mediated autophagy regulation, ferroptosis, and the role of apolipoprotein M (ApoM) in preserving lysosomal integrity. Additionally, we discuss how impaired lysosomal acidification, mediated by V-ATPase, contributes to lipid-induced cardiac dysfunction. Therapeutically, several pharmacological agents, such as statins, SGLT2 inhibitors, TRPML1 agonists, resveratrol, curcumin, and ferroptosis modulators (e.g., GLS1 activators and icariin), have demonstrated promise in restoring lysosomal function, enhancing autophagic flux, and reducing inflammatory and oxidative injury in both experimental models and early clinical settings. However, key challenges remain, including limitations in drug delivery systems, the absence of lysosome-specific biomarkers, and insufficient clinical validation of these strategies. Future research should prioritize the development of reliable diagnostic tools for lysosomal dysfunction, the optimization of targeted drug delivery, and large-scale clinical trials to validate therapeutic efficacy. Incorporating lysosome-modulating approaches into standard cardiovascular care may offer a new precision medicine paradigm for managing CVD progression.
Keywords: lysosomal stress; statins; transcription factor EB; glutaminase 1; senescence; NLRP3 inflammasome; SGLT2 inhibitors; regulatory complex; trehalose
1. Introduction
Cardiovascular diseases (CVDs) remain the leading cause of morbidity and mortality worldwide and are primarily driven by metabolic disorders, such as obesity, type 2 diabetes mellitus, and atherosclerosis [1]. These conditions accelerate CVD progression through chronic inflammation, oxidative stress, and metabolic dysregulation, ultimately resulting in complications such as chronic cardiomyopathy, pressure overload, and heart failure (HF) [2]. Metabolic dysfunction contributes to various forms of organelle stress, including endoplasmic reticulum stress, mitochondrial dysfunction, and lysosomal stress, all of which collectively disrupt cellular homeostasis and promote vascular inflammation [3–5]. Ref. [6] notes the disruption of lysosomal homeostasis, and Ref. [7] further emphasizes the promotion of vascular inflammation and atherosclerosis, which are crucial for the pathogenesis of cardiovascular complications.
Biomedicines 2025, 13, 1053 https://doi.org/10.3390/biomedicines13051053
Lysosomes are essential organelles responsible for the degradation and recycling of cellular waste via autophagy and endocytosis [8]. However, emerging evidence has expanded the understanding of lysosomes beyond their conventional roles, revealing them as dynamic signaling hubs regulating various cellular processes, including the mechanistic target of the mammalian target of rapamycin complex 1 (mTORC1), 5′ adenosine monophosphateactivated protein kinase (AMPK), and inflammasome signaling pathways [9,10]. Additionally, lysosomes contribute to extracellular processes, such as microcirculation, through secretory lysosomes, underscoring their systemic impact on cellular and tissue health.
Lysosomes are frequently subjected to damage due to various internal and external factors [11]. When damaged, they release protons and cathepsins, leading to cell death and inflammation [11]. Recent studies have shown that cells possess a collective defense mechanism known as the lysosomal damage response to mitigate the effects of lysosomal injury. This response includes lysophagy, a form of selective autophagy; membrane repair pathways mediated by endosomal sorting complexes required for protein transport; and biosynthetic pathways regulated by transcription factor EB (TFEB). These mechanisms function synergistically to maintain lysosomal homeostasis [12].
Lysosomal dysfunction has been implicated in rare genetic conditions, such as lysosomal storage diseases, and prevalent age-related disorders, including cardiovascular and neurodegenerative diseases [13,14]. The concept of “lysosomal stress” has recently emerged, emphasizing its critical role in driving chronic inflammation, oxidative stress, and metabolic dysregulation, which are significant contributors to CVD progression [15]. This emerging evidence suggests that lysosomes are promising therapeutic targets for addressing the mechanisms underlying CVDs.
Recent advances in cardiovascular drugs have led to the investigation of strategies that target lysosomal pathways to mitigate inflammation, oxidative stress, and metabolic dysfunction [16]. By modulating lysosomal activity, these therapies aim to restore cellular homeostasis, reduce the progression of atherosclerosis, and stabilize vulnerable plaques. This review explores the role of lysosomal dysfunction in CVDs and discusses emerging cardiovascular drugs that leverage lysosome-targeted approaches to improve clinical outcomes.
2. Lysosomal Dysfunction, NLRP3 Inflammasome, and CVDs:Mechanisms and Therapeutic Insights
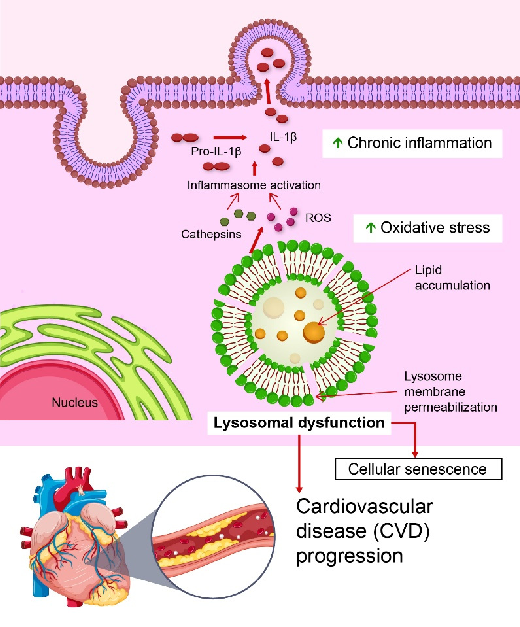
Lysosomal dysfunction, particularly lysosomal rupture, is vital for the pathogenesis of CVDs. Lysosomal rupture involves the complete loss of lysosomal membrane integrity, resulting in the uncontrolled release of cathepsins into the cytosol [17]. This process triggers potent inflammatory responses, including the activation of the NOD, LRR, and pyrin domain-containing protein 3 (NLRP3) inflammasome, which is a crucial mediator of chronic inflammation in CVDs (Figure 1). Latz et al. first demonstrated that cholesterol crystals internalized by macrophages in atherosclerotic plaques induce lysosomal rupture, releasing cathepsin B and driving NLRP3 activation, leading to sustained vascular inflammation [18].
Similarly, Karasawa et al. showed that saturated fatty acids, such as palmitic acid, induce intracellular crystal formation, causing lysosomal damage and activating the NLRP3 inflammasome in macrophages [19]. Conversely, unsaturated fatty acids, such as oleic acid, prevent this process by inhibiting crystal formation, underscoring the therapeutic significance of maintaining the balance between saturated and unsaturated fatty acids [19].
In contrast, lysosomal membrane permeabilization (LMP) represents a partial and controlled disruption of lysosomal membranes that regulates NLRP3 inflammasome activity under specific conditions [20]. For example, in human vascular smooth muscle cells, LMP acts as an activation signal linking lysosomal dysfunction to vascular inflammation [21].
controlled disruption of lysosomal membranes that regulates NLRP3 inflammasome activity under specific conditions [20]. For example, in human vascular smooth muscle cells,
These insights highlight the potential of targeting lysosomal integrity and inflammasome regulation as therapeutic strategies for CVD.
[21]. These insights highlight the potential of targeting lysosomal integrity and inflammasome regulation as therapeutic strategies for CVD.
Figure 1. Role of lysosomal dysfunction in the progression of cardiovascular disease (CVD). Lysosomal dysfunction, characterized by lysosomal membrane permeabilization and lipid accumulation, leads to the release of cathepsins and reactive oxygen species (ROS). These factors activate inflammasomes, promoting the cleavage of pro-IL-1β into its active form, IL-1β, which triggers chronic inflammation. In addition, oxidative stress exacerbates lysosomal damage [22]. These processes collectively drive cellular senescence and contribute to CVD progression. This figure highlights the interplay between lysosomal dysfunction, chronic inflammation, and oxidative stress in
- 2.1. Cardiovascular Drugs Targeting Lysosomal Stress and NLRP3 Inflammasome Activation
Several cardiovascular drugs have demonstrated the potential to mitigate lysosomal stress and associated inflammation by restoring lysosomal function and suppressing NLRP3 inflammasome activation.
- 2.1. Cardiovascular Drugs Targeting Lysosomal Stress and NLRP3 Inflammasome Activation
Several cardiovascular drugs have demonstrated the potential to mitigate lysosomal stress and associated inflammation by restoring lysosomal function and suppressing NLRP3 inflammasome activation.
- 2.1.1. Statins
- 2.1.2. SGLT2 Inhibitors
- 2.1.1. Statins
In addition to their lipid-lowering effects, HMG-CoA reductase inhibitors (statins) have been shown to suppress NLRP3 inflammasome activation through PPAR-γ activation, highlighting their potential anti-inflammatory benefits beyond cholesterol reduction [23]. By inducing autophagy, simvastatin promotes the clearance of damaged lysosomes,
Dapagliflozin, a sodium–glucose co-transporter-2 (SGLT2) inhibitor, has demonstrated cardioprotective effects beyond glucose lowering [25,26]. In a mouse model of myocardial ischemia/reperfusion (I/R) injury, dapagliflozin reduced infarct size, cardiac damage
markers, and inflammation by suppressing NLRP3 inflammasome activation. These effects are mediated by enhanced autophagy and improved lysosomal function, which promotes NLRP3 clearance [27]. In primary cardiomyocytes, dapagliflozin reduced intracellular Ca2+ and Na+ levels, further supporting its role in protecting against I/R injury.
- 2.1.3. Vitamin E
In vivo, Vitamin E predominantly exists in its free form, which enables it to exert antioxidative effects more efficiently than its esterified form [28]. Clinical studies indicate that a minimum of 100 IU of Vitamin E is required to inhibit LDL oxidation [29], a key process in the development of atherosclerosis and CVD. Several clinical trials, including the Cambridge Heart Antioxidant Study (400–800 IU/day) and the SPACE trial (800 IU/day), have reported the cardioprotective effects of Vitamin E [30,31]. However, other trials, such as GISSI [32], HOPE [33], and PPP [34], have failed to confirm its efficacy, contributing to an ongoing debate regarding the role of oxidative stress in CVD. Levy et al. suggested that individuals with reduced antioxidative capacity may derive greater benefits from Vitamin E supplementation [35].
- 2.2. Regulatory Complex and CVD
The regulatory complex of lysosomal membrane proteins plays a role in linking lysosomal metabolic signaling pathways with immune response regulation mechanisms [36]. The regulator complex, located on the lysosomal membrane, is composed of Lamtor2 (p14), Lamtor3 (MP10), Lamtor4 (p10), and Lamtor5 (HBXIP), which are encapsulated by Lamtor1 (p18) [37]. Among these components, Lamtor1 is indispensable for maintaining the overall stability of the regulatory complex. The regulator complex primarily functions in metabolic signaling by anchoring mTORC1 to the lysosomal membrane, integrating intracellular nutrient signals, and regulating protein and lipid synthesis and degradation. Kimura et al. reported that the Lamtor1-mTORC1 pathway transmits signals to liver X receptors (LXRs) based on the macrophage environment and nutrient status, thereby modulating the activation of M2 (anti-inflammatory) macrophages [38]. Hayama et al. demonstrated that the regulation of TFEB via the Lamtor1-mTORC1 pathway is involved in lysosomal autophagy and the production of inflammatory cytokines [39]. Recent advances, including the findings of Evavold et al., have indicated that the Lamtor1-mTORC1 pathway promotes the production of mitochondrial ROS and subsequent pyroptosis through gasdermin D (GSDMD) pore formation. These studies underscore the role of the regulator complex in linking lysosomal metabolic signaling pathways with immune response regulation mechanisms. Emerging research has highlighted the regulatory complex as a mediator of NLRP3 inflammasome activation. Lamtor1, a regulatory complex component, interacts with NLRP3 and histone deacetylase 6 (HDAC6) to modulate inflammasome activity. Lamtor1 deletion in macrophages significantly reduced NLRP3 activation, demonstrating its regulatory role in inflammation. Furthermore, DL-all-rac-α-tocopherol, a synthetic form of Vitamin E, inhibits the interaction between Lamtor1 and HDAC6, reducing NLRP3 activation and alleviating inflammation in gouty arthritis models [40]. Vitamin E has also been associated with a reduction in cardiovascular events [30,31,35]. However, while evidence supports the role of DL-all-rac-α-tocopherol in inhibiting Lamtor1-HDAC6 interactions and reducing NLRP3 activation under inflammatory conditions, its effects on cardiovascular disease remain inconclusive. Specifically, it has not been definitively established whether Vitamin E exerts the same inhibitory effects in cardiovascular pathology. The direct link between Vitamin E-mediated inhibition of Lamtor1-HDAC6 interactions, reduced NLRP3 activation, and the prevention of cardiovascular events remains unverified. Future studies are needed
to clarify whether these mechanisms contribute to the cardioprotective effects of Vitamin E and its potential as a therapeutic target for cardiovascular disease.
- 2.3. Uric Acid, Lysosomal Dysfunction, and NLRP3 Inflammasome Activation in Cardiovascular Disease
- 2.4. PCSK9 Inhibition and Its Potential Impact on Lysosomal Function
Although PCSK9 inhibitors have been widely studied for their role in cholesterol regulation and atherosclerosis management, their effects on lysosomal function remain largely unexplored [51]. Various forms of PCSK9 inhibitors, including monoclonal antibodies (mAbs), small interfering RNAs (siRNAs), and vaccines, have demonstrated clinical efficacy in lowering LDL cholesterol levels and reducing cardiovascular risk [52–54]. Recent findings suggest that PCSK9 interacts with the NLRP3 inflammasome, potentially influencing the inflammatory processes involved in atherosclerosis progression [55,56]. However, whether PCSK9 inhibition affects lysosomal integrity, autophagy regulation, or lipid processing within lysosomes is yet to be fully established [57,58]. Given the fundamental role of lysosomes in cholesterol metabolism and immune signaling, further investigation is required to determine whether PCSK9 inhibition modulates lysosomal stability, autophagic flux, or NLRP3 inflammasome activation via lysosomal pathways [59,60]. Clarifying these mechanisms could lead to novel therapeutic strategies that integrate PCSK9 inhibitors with lysosome-targeting therapies to optimize the treatment of atherosclerosis, metabolic disorders, and inflammatory CVDs [61,62].
3. TFEB: A Key Player in Lysosomal Stress and Cardiovascular Therapy
Lysosomal dysfunction plays a critical role in the progression of CVDs by contributing to chronic inflammation, oxidative stress, and impaired metabolic regulation. Recent research has identified TFEB as a central regulator of lysosomal homeostasis, making it a key target for therapeutic intervention in CVDs [63]. TFEB functions as a master transcrip-
tional regulator, coordinating lysosomal biogenesis, autophagy, and cellular adaptation to metabolic stress [64]. Given its essential role in maintaining cellular quality control, TFEB activation is increasingly being recognized as a promising strategy for mitigating cardiovascular complications.
One of the primary mechanisms by which TFEB exerts its protective effects is through the enhancement of lysosomal biogenesis and function [65]. By upregulating genes involved in lysosomal stability and acidification, TFEB ensures the efficient degradation of cellular debris and prevents lysosomal rupture, which is a key driver of inflammatory responses in atherosclerosis and HF. Furthermore, TFEB activation supports autophagy, which is essential for the removal of damaged mitochondria and lipid-laden foam cells, thereby reducing vascular inflammation and plaque instability [66]. Recent studies have suggested that TFEB also plays a crucial role in mitigating oxidative stress, which is a major contributor to endothelial dysfunction and cardiomyocyte apoptosis [67–69]. By regulating lysosomal repair pathways, TFEB helps maintain redox balance, which is essential for preserving cardiovascular function under conditions of metabolic and hemodynamic stress. As the molecular pathways governing TFEB activation become better understood, there is a growing interest in leveraging this transcription factor in cardiovascular therapy [70,71]. The following sections discuss the involvement of TFEB in atherosclerosis, its role in vascular smooth muscle cell stability, and the therapeutic potential of pharmacological agents in enhancing TFEB activity.
- 3.1. Role of TFEB in Atherosclerosis and Endothelial Damage
Emanuel et al. [19] demonstrated that oxidized low-density lipoproteins and cholesterol crystals induce lysosomal dysfunction in macrophages. TFEB overexpression in these cells restores lysosomal function, promotes autophagy, and reduces inflammation, highlighting its therapeutic potential in vascular inflammation and atherosclerosis [72].
- 3.2. TFEB in Vascular Smooth Muscle Cells and Plaque Stability
Vascular smooth muscle cells (VSMCs) are significant contributors to plaque stability, partly through the production of cystathionine gamma-lyase (CTH), which generates hydrogen sulfide (H2S), a protective gasotransmitter in atherosclerosis. Reduced CTH expression in VSMCs has been associated with impaired autophagy, lysosomal dysfunction, and increased plaque vulnerability [25]. Experimental studies have demonstrated that Cth deletion in VSMCs exacerbates lysosomal and autophagic deficits, increases apoptosis, and reduces collagen secretion, leading to plaque instability [73]. H2S donors have been shown to rescue these effects by activating TFEB, a master regulator of autophagy and lysosomal function. H2S sulfhydrates TFEB at Cys212 and promotes its nuclear translocation and transcriptional activation of lysosomal and autophagy-related genes, such as ATG9A and LAPTM5. This activation restores lysosomal function, enhances autophagic flux, supports collagen production, and stabilizes plaques. These findings underscore the significance of the CTH-H2S-TFEB axis in maintaining lysosomal integrity and autophagy in VSMCs, offering promising therapeutic and biomarker potential for vulnerable plaques.
- 3.3. Cardiovascular Drugs Enhancing TFEB Activity
- 3.3.1. Statins
Simvastatin: This promotes TFEB activation by inhibiting mTORC1, activating AMPK, enhancing autophagy, and reducing lipid accumulation in atherosclerotic models [74].
Atorvastatin: It increases TFEB protein levels in cardiac tissue and improves lysosomal function in doxorubicin-induced cardiomyopathy, as indicated by an enhanced lysosomal membrane protein (LAMP)2/tubule-associated protein 1 light chain 3 beta (LC3B) ratio [75].
- 3.3.2. TRPML1 Agonists
- 3.3.3. Trehalose
- 3.3.4. Resveratrol (RSV)
- 3.3.5. Curcumin (Cur)
Cur, a polyphenol derived from Curcuma longa, has been shown to mitigate atherosclerosis by promoting TFEB activation, enhancing autophagy, and reducing inflammation [85]. It facilitates the nuclear translocation of TFEB and improves lysosomal biogenesis and lipid catabolism in foam cells, thereby preventing excessive lipid accumulation. Beyond its role in autophagy, curcumin suppresses inflammation by inhibiting the P300-BRD4 axis, which is involved in the transcriptional activation of pro-inflammatory genes in atherosclerosis. This dual mechanism not only enhances lipid clearance but also reduces oxidative stress and inflammatory cytokine production. In ApoE-deficient mouse models, curcumin administration reduces atherosclerotic plaque formation, improves vascular integrity, and restores autophagic flux. These effects depend on TFEB activation, highlighting the potential of curcumin as a therapeutic agent for lysosomal dysfunction in CVDs.
- 3.3.6. Traditional Chinese Medicine: Dehydroandrographolide (DA)
DA, an active component of Andrographis paniculata, mitigates doxorubicin-induced cardiotoxicity by enhancing lysosomal function and autophagic flux. DA inhibits mTOR activity and promotes TFEB nuclear translocation [86]. Although these findings highlight the potential of DA in repairing lysosomal dysfunction in CVDs, its application remains limited to experimental models.
- 3.4. Eicosapentaenoic Acid (EPA) and Lysosomal Homeostasis
Large-scale randomized clinical trials, including the REDUCE-IT [87] and STRENGTH [88] trials, have investigated the cardiovascular benefits of high-dose omega-3 polyunsaturated fatty acids (PUFAs). Notably, REDUCE-IT reported a 25% reduction in cardiovascular events, whereas STRENGTH showed only a 1% risk reduction, leading to conflicting interpretations regarding whether EPA truly reduced cardiovascular risk. Subsequent cohort studies have suggested that differences in the comparator oils used in these trials may partially explain the disparity in outcomes [89]; however, the overall efficacy of EPA remains uncertain.
Beyond its established roles in lipid metabolism, inflammation reduction, and endothelial function improvement, emerging evidence suggests that EPA may also influence lysosomal function, which is a key component of cellular homeostasis. Lysosomes facilitate the degradation of damaged organelles and proteins via autophagy, a process essential for cardiovascular protection. Lysosomal dysfunction contributes to CVDs, including ischemia/reperfusion injury and atherosclerosis, by impairing cellular clearance mechanisms and exacerbating oxidative stress. Several studies have indicated that EPA promotes autophagy, facilitating adaptive cellular responses that counteract oxidative stress-induced cardiomyocyte apoptosis [90]. However, whether this effect is mediated through the direct modulation of lysosomal pathways remains unclear. Given the fundamental role of lysosomes in cholesterol metabolism, inflammatory regulation, and cellular clearance, further research is required to determine the precise effects of EPA on lysosomal integrity, autophagic flux, and metabolic balance. Future investigations should explore whether EPA influences key lysosomal regulators, such as TFEB or TRPML1, to provide valuable insights into its role in lysosome-targeted therapies. A deeper understanding of the interactions between EPA and lysosomal biology may lead to novel therapeutic strategies for CVDs associated with lysosomal dysfunction and impaired autophagy.
4. Ferroptosis in Cardiovascular Disease: From Plaque Destabilization toMyocardial Injury
Ferroptosis, an iron-dependent form of cell death, plays a crucial role in the destabilization of atherosclerotic plaques and the progression of CVDs. Metabolic disorders, such as obesity, diabetes, and atherosclerosis, promote ferroptosis by increasing oxidative stress, lipid peroxidation, and lysosomal dysfunction, thereby exacerbating cardiovascular risk [91–94]. Lysosomal dysfunction contributes to this process by releasing iron and ROS, which amplifies ferroptosis in VSMCs and increases plaque vulnerability. Recent studies have further highlighted metabolic reprogramming in unstable plaques with intraplaque hemorrhage (IPH+), revealing impaired glutamine-to-glutamate conversion and reduced glutamate levels. These metabolic alterations are associated with an increased presence of macrophages and a pro-inflammatory microenvironment. The interplay between metabolic dysfunction and ferroptosis exacerbates plaque instability, underscoring the therapeutic potential of targeting lysosomal dysfunction and metabolic pathways in CVDs [95].
A recent study revealed that the Yes-associated protein 1 (YAP1)/glutaminase 1 (GLS1) axis is a significant regulator of ferroptosis in VSMCs [96]. GLS1, an enzyme essential for
glutamine metabolism, enhances the production of glutamate and glutathione, which are critical for maintaining redox balance and preventing lipid peroxidation. YAP1 signaling activates GLS1, mitigates ferroptosis, stabilizes VSMCs, and reinforces atherosclerotic plaques. Conversely, GLS1 inhibition exacerbates oxidative stress and plaque instability, suggesting that enhancing GLS1 activity could serve as a novel therapeutic strategy to counteract ferroptosis in CVDs.
- 4.1. Cardiovascular Drugs Targeting Ferroptosis and Lysosomal Stress
- 4.1.1. GLS1 Activators
Pharmacological agents targeting GLS1 or promoting glutathione synthesis complement statins by directly reducing ferroptosis in VSMCs and stabilizing plaques [96]. Combining statins with GLS1 activators has potential synergistic effects on plaque stability and overall cardiovascular outcomes.
- 4.1.2. Icariin Icariin, derived from Epimedium brevicornum Maxim., has demonstrated protective effects against ferroptosis in vascular endothelial cells exposed to oxidized low-density lipoproteins [97]. It reduces ROS levels, promotes TFEB nuclear translocation, enhances autophagosome–lysosome fusion, increases autophagy, and decreases ferroptosis. In highfat diet-fed ApoE-deficient mice, icariin alleviated atherosclerotic lesions, demonstrating its potential as a candidate for CVD treatment by targeting lysosomal pathways and ferroptosis.
- 4.1.3. Cur
- 4.1.4. Other Therapeutic Approaches
- 5. Future Challenges in Bridging Experimental and Clinical Research on Lysosomal Dysfunction
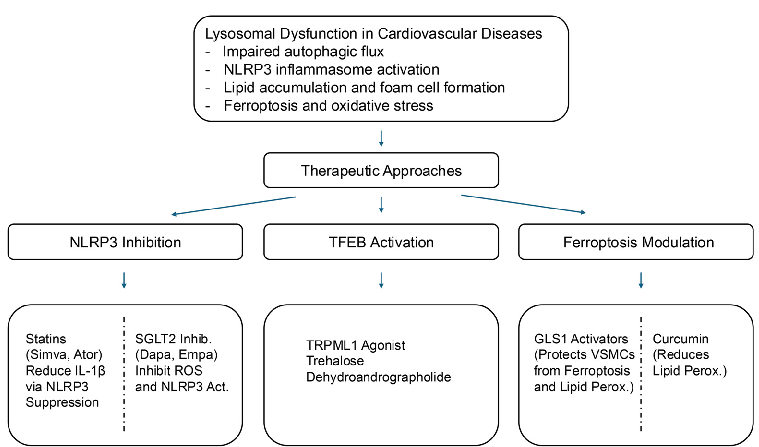
We summarize the therapeutic strategies targeting lysosomal dysfunction in cardiovascular diseases in Figure 2, which highlights key pharmacological agents and their corresponding molecular targets. Lysosomal dysfunction, NLRP3 inflammasome activation, and ferroptosis have emerged as critical mechanisms in the pathogenesis of CVDs, including atherosclerosis, HF, and metabolic syndrome [18,19,41,101,102]. Although significant progress has been made in understanding these pathways, several challenges must be addressed to translate these findings into effective clinical therapies.
Biomedicines 2025, 13, 1053 progress has been made in understanding these pathways, several challenges must be10 of 23 addressed to translate these findings into effective clinical therapies.
Figure 2. Therapeutic strategies targeting lysosomal dysfunction in cardiovascular diseases. This schematic illustrates lysosomal dysfunction as a central contributor to CVDs and highlights emerging therapeutic strategies targeting these pathways. Lysosomal stress disrupts autophagic flux, promotes lipid accumulation, activates the NLRP3 inflammasome, and triggers ferroptosis, collectively exacerbating inflammation and oxidative stress.
- 5.1. Advancing Lysosome-Targeted Therapies
- 5.2. Overcoming Challenges in NLRP3 Inflammasome Inhibition
- 5.1. Advancing Lysosome-Targeted Therapies
- 5.2. Overcoming Challenges in NLRP3 Inflammasome Inhibition
Inflammation is a key driver of CVD progression, and NLRP3 inflammasome activation plays a central role in this process. Pharmacological inhibitors of the NLRP3 inflammasome, such as statins, SGLT2 inhibitors, and Vitamin E, have demonstrated anti-inflammatory and cardioprotective effects. However, the precise mechanisms underlying the modulation of lysosomal function remain unclear. Further research is needed to determine whether lysosomal integrity directly regulates inflammasome activation in cardiovascular pathology and how these interventions can be optimized for long-term use.
Inflammation is a key driver of CVD progression, and NLRP3 inflammasome activation plays a central role in this process. Pharmacological inhibitors of the NLRP3
Emerging evidence suggests that UA crystals can activate the NLRP3 inflammasome through lysosomal rupture, contributing to tissue damage in hyperuricemia-related CVDs [41–43]. Future studies should explore how targeting UA metabolism and lysosomal integrity can reduce inflammation and improve CVD outcomes.
- 5.3. Addressing the Role of Ferroptosis in Cardiovascular Disease
- 5.4. Future Perspectives and Translational Research
To successfully implement lysosome-targeted therapies in CVD treatment, future research should prioritize the following:
- 1. Clinical Trials and Drug Optimization: Expanding human trials for lysosomemodulating drugs, including TFEB activators, TRPML1 agonists, and ferroptosis inhibitors, is crucial for clinical translation.
- 2. Personalized Medicine Approaches: Identifying patient subgroups with heightened lysosomal stress or inflammasome activation could allow for targeted interventions tailored to specific cardiovascular conditions.
- 3. Biomarker Development: Establishing reliable biomarkers for lysosomal dysfunction, NLRP3 activation, and ferroptosis will aid in disease diagnosis, treatment monitoring, and therapy selection.
By addressing these challenges, lysosome-targeted interventions have the potential to revolutionize CVD treatment, offering a novel approach for mitigating chronic inflammation, oxidative stress, and metabolic dysfunction associated with cardiovascular pathology.
Key therapeutic strategies include the following:
- • NLRP3 Inflammasome Inhibition: Statins, SGLT2 inhibitors, and Vitamin E suppress NLRP3 activation, thereby reducing inflammation.
- • TFEB Activation: Statins, TRPML1 agonists, trehalose, resveratrol, curcumin, and dehydroandrographolide (DA) enhance lysosomal biogenesis and autophagic clearance.
- • Ferroptosis Modulation: GLS1 activators, icariin, and curcumin, regulate irondependent cell death, thereby protecting against cardiovascular damage.
- • Lysosomal Repair Mechanisms: TRPML1 activation and lysophagy modulation stabilize lysosomal membranes and restore function.
By restoring lysosomal homeostasis, these strategies may reduce oxidative stress and improve cardiovascular outcomes. Further clinical validation is required.
6. Lysosomal Dysfunction and Cellular Senescence: Implications forCardiovascular Health
Cellular senescence is a state of irreversible cell cycle arrest induced by stressors such as DNA damage, oxidative stress, and oncogenic signals [105]. Despite their inability to divide, senescent cells remain metabolically active and secrete various inflammatory and matrix-degrading factors, referred to as the senescence-associated secretory phenotype (SASP) [106,107]. The SASP promotes chronic inflammation and tissue remodeling, significantly contributing to the development of CVDs and obesity-related metabolic disorders [108].
Recent evidence suggests that eliminating senescent cells alleviates age-related conditions, including atherosclerosis and chronic kidney disease, while extending the health span of animal models [109]. For example, selective clearance of senescent cells in transgenic mice reduces atherosclerotic plaque size, improves cardiac function, and mitigates systemic inflammation. These findings have prompted the development of senotherapy, a therapeutic approach aimed at targeting senescent cells to treat or prevent aging-related diseases. Several of these senotherapeutics have advanced to clinical trials, providing potentially promising treatment options for age-related conditions and disorders, including atherosclerosis, cancer, diabetes, neurodegenerative diseases, CVDs, and chronic kidney disease [110].
- 6.1. Lysosomal Dysfunction in Cellular Senescence
- 6.2. Cardiovascular Drugs Targeting Senescence via Lysosomal Pathways
- 6.3. Targeting GLS1 for Cardiovascular Benefits
Recent studies have identified GLS1 as a novel target for immunotherapy [113]. In senescent cells, lysosomal damage induces intracellular acidification, stabilizes GLS1 mRNA, and enhances GLS1 protein expression. This mechanism increases ammonia production, neutralizes acidification, and supports the survival of senescent cells. GLS1 inhibition disrupts this compensatory mechanism, exacerbates intracellular acidification, and induces senescence. Preclinical studies have shown that GLS1 inhibitors can effectively clear senescent cells, reduce inflammation, and improve metabolic and cardiovascular health.
7. Lysosomal Stress and the Role of V-ATPase in Lipid-InducedCardiac Dysfunction
Lysosomal stress has been identified as a critical factor in the pathogenesis of diabetic cardiomyopathy [114]. Lipid overload disrupts vacuolar H+-ATPase (V-ATPase) function, which is a key component of lysosomal and endosomal homeostasis, leading to endosomal deacidification. This disruption results in the aberrant trafficking of CD36, a lipid transporter, from the endosomes to the sarcolemma, exacerbating lipid accumulation, insulin resistance, and cardiac contractile dysfunction.
Wang et al. further demonstrated the therapeutic potential of specific amino acids (lysine, leucine, arginine) in mitigating lysosomal stress [115]. These amino acids reassembled V-ATPase, restored lysosomal and endosomal homeostasis, and reduced lipid accumulation in lipid-overloaded cardiomyocytes and high-fat diet-fed rats. The observed effects relied on the mTORC1-V-ATPase axis, adapter protein Raptor, and lysosomal amino acid transporter SLC38A9. These findings highlight lysosomal stress as a pivotal mechanism linking metabolic dysfunction and cardiovascular disease, underscoring the potential of targeting the V-ATPase-mTORC1 axis using specific amino acids as a novel therapeutic approach.
8. Apolipoprotein M (ApoM) and Lysosomal Function inCardiovascular Disease
Clinical studies have indicated that reduced circulating ApoM is associated with increased mortality in patients with HF, independent of the HF subtype, ischemic heart disease, or HDL cholesterol levels [116,117]. ApoM, which binds to sphingosine-1-phosphate (S1P), is negatively affected by diabetes, particularly in HFpEFs. Recent findings suggest that anthracyclines, such as doxorubicin (Dox), reduce circulating ApoM levels in both mice and humans, whereas ApoM heterozygosity exacerbates DOX-induced cardiotoxicity [118]. Mechanistically, ApoM appears to sustain myocardial autophagic flux, protect against lysosomal injury, and mitigate DOX-induced cardiotoxicity. Studies using anthracyclineinduced HF models have identified ApoM as a novel regulator of the autophagy–lysosomal pathway, highlighting its potential therapeutic role in HF management.
9. Conclusions
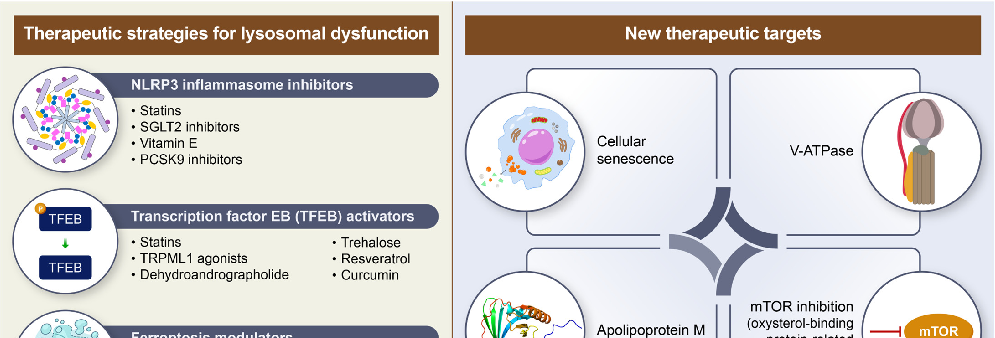
Integrating therapies targeting lysosomal dysfunction, cellular senescence, and ferroptosis into existing CVD treatment regimens has great potential for improving patient outcomes (Figure 3).
9.1. Pharmacological Approaches
Several pharmacological strategies are being explored to target lysosomal dysfunction in cardiovascular diseases. One key approach involves inhibiting the NLRP3 inflammasome, a major driver of inflammation and oxidative stress. Statins, SGLT2 inhibitors, and Vitamin E have demonstrated the ability to suppress NLRP3 activation, thereby reducing inflammatory responses and mitigating oxidative damage.
Another promising avenue focuses on activating TFEB, a transcription factor critical for lysosomal biogenesis and autophagy. Pharmacological agents such as statins, TRPML1 agonists, trehalose, resveratrol, curcumin, and dehydroandrographolide have been shown to enhance lysosomal function, thereby alleviating lipid overload and chronic inflammation.
Modulating ferroptosis, an iron-dependent form of cell death, is also emerging as a therapeutic target. Ferroptosis contributes to cardiovascular damage by disrupting lysosomal integrity and metabolic balance. Agents such as GLS1 activators, icariin, and curcumin have been investigated for their ability to regulate ferroptotic pathways and protect against cardiovascular dysfunction.
BiomedicinesBiomedicines20252025, 13,, 105313, x FOR PEER REVIEW 14 of 2514 of 23
Figure 3. Therapeutic strategies targeting lysosomal dysfunction in CVDs. This schematic illustrates the key therapeutic strategies designed to mitigate lysosomal dysfunction in CVDs by targeting various pathological mechanisms.
Figure 3. Therapeutic strategies targeting lysosomal dysfunction in CVDs. This schematic illustrates the key therapeutic strategies designed to mitigate lysosomal dysfunction in CVDs by targeting various pathological mechanisms.
9.2. Emerging Therapeutic Targets
- 9.1. Pharmacological Approaches
- 9.2. Emerging Therapeutic Targets
Beyond these established pharmacological approaches, novel strategies aimed at lysosomal dysfunction in cardiovascular diseases are gaining attention. One such area of interest is cellular senescence, where interventions targeting age-related lysosomal decline could help prevent cardiovascular aging and dysfunction.
Regulating lysosomal acidification through V-ATPase modulation has also been proposed as a strategy to restore autophagic flux and improve lipid degradation. Additionally, apolipoprotein M (ApoM) has been identified as a key player in maintaining endothelial lysosomal function through its influence on sphingosine-1-phosphate signaling, which is essential for vascular homeostasis.
Another potential target is the regulation of mTOR activity through ORP5-related pathways. The oxysterol-binding protein-related protein 5 (ORP5) has been implicated in mTORC1 regulation within lysosomes, influencing cardiovascular metabolism and contributing to conditions such as hypertrophy.
While these therapeutic strategies show promise, further clinical research is necessary to validate their efficacy and safety. Large-scale trials will be critical in determining their potential for integration into existing cardiovascular treatment regimens. A comprehensive table summarizing the key drugs discussed in this review, along with their mechanisms of action and clinical status, is provided below (Table 1).
Beyond these established pharmacological approaches, novel strategies aimed at lysosomal dysfunction in cardiovascular diseases are gaining attention. One such area of interest is cellular senescence, where interventions targeting age-related lysosomal decline could help prevent cardiovascular aging and dysfunction.
Continued research in these areas is likely to yield innovative therapeutic strategies that address the underlying pathophysiology of CVDs and extend the health span of the aging population.
Regulating lysosomal acidification through V-ATPase modulation has also been pro-
Table 1. Mechanism of action and clinical status of lysosome-targeted therapies.
Drug Class Mechanism of Action Clinical Status Key Clinical Findings NLRP3 Inflammasome Inhibitors
Inhibits NLRP3 inflammasome via AMPK activation, reducing inflammation
Lowers inflammatory burden in CVD
Statins
Clinical use
Suppresses NLRP3 inflammasome activation enhances autophagy
Reduces myocardial infarction size, cardiac damage markers
SGLT2 Inhibitors
Clinical use
Inhibits Lamtor1-HDAC6 interaction, reducing NLRP3 activation
Associated with reduced CVD risk Cardiovascular Drugs Enhancing TFEB Activity Statins
Observational and preclinical studies
Vitamin E
Enhances TFEB activity via mTORC1 inhibition, AMPK activation
Improves lysosomal function, reduces lipid accumulation
Widely used in clinical practice
Activates TFEB via lysosomal calcium signaling
Protects against oxidative stress and autophagic defects
TRPML1 Agonists
Preclinical
Promotes TFEB activation, enhances lysosomal biogenesis
Preclinical, under investigation
Reduces plaque burden, enhances autophagy
Trehalose
Stimulates ER-Ca2+ signaling and activates TFEB
Improves lipid metabolism and autophagy regulation
Preclinical evidence
Resveratrol (RSV)
Reduces foam cell formation and inflammation
Promotes TFEB activation, enhances lipid catabolism
Curcumin (Cur)
Preclinical studies
Dehydroandrographolide (DA)
Activates TFEB enhances the lysosomal function
Improves autophagic flux, reduces CVD progression
Experimental models
Ferroptosis Modulators
Prevents oxidative stress-induced VSMC death
Enhances glutaminolysis, reducing ferroptosis
GLS1 Activators
Preclinical studies
Reduces ROS, promotes TFEB nuclear translocation, prevents ferroptosis
Reduces atherosclerotic lesions
Icariin
Preclinical models
10. Future Directions in Lysosome-Targeted Cardiovascular Therapies
Lysosomal dysfunction plays a pivotal role in the pathogenesis of CVDs; however, its clinical significance remains underexplored. While preclinical studies have established that lysosomal stress contributes to inflammation, oxidative stress, and metabolic dysregulation in CVD, translating these findings into therapeutic applications remains challenging [9,11]. Future research should focus on refining lysosome-targeted strategies, integrating them into existing treatment regimens and developing clinical approaches for severe CVD, including HF [14,34]. Key future research directions in lysosome-targeted cardiovascular therapies are summarized in Table 2.
- 10.1. Advancing Lysosomal-Targeted Pharmacological Therapies
- 10.2. Clinical Trials on Lysosome-Targeted Therapies in CVD
- 10.3. Exploring Novel Biomarkers and Diagnostic Tools for Lysosomal Dysfunction
- 10.4. Translating Lysosome-Targeted Strategies into Clinical Practice
Lysosomal dysfunction has emerged as a key contributor to the progression of severe CVDs, particularly HF. Despite growing recognition of its importance, the direct clinical implications of lysosomal dysfunction in HF remain underexplored. Given that lysosomes regulate autophagy, metabolic homeostasis, and inflammatory signaling, their impairment can accelerate myocardial remodeling, fibrosis, and contractile dysfunction, thereby exacerbating HF progression. Understanding how lysosomal pathways influence these
pathological mechanisms is crucial for the development of effective therapeutic strategies. Recent findings have identified mTORC1 activation in lysosomes as a key regulator of metabolic adaptations in the failing heart. Dysregulated mTORC1 signaling has been implicated in the pathogenesis of cardiac hypertrophy and metabolic inflexibility, both of which contribute to HF. In this context, oxysterol-binding protein-related protein 5 (ORP5) has been found to localize to lysosomal membranes, where it promotes mTORC1 activation, leading to enhanced anabolic signaling and metabolic stress. Increased ORP5 expression has been associated with pathological cardiac hypertrophy, suggesting that aberrant ORP5-mTORC1 interactions may drive maladaptive metabolic responses in HF [119]. These findings indicate that targeting ORP5-mediated mTORC1 activation may represent a novel therapeutic approach for mitigating myocardial stress and preserving cardiac function. Translating lysosomal-targeted strategies into clinical practice requires further investigation to determine whether pharmacological interventions can restore lysosomal integrity and improve cardiac outcomes in patients with HF. TFEB activators, TRPML1 agonists, and NLRP3 inflammasome inhibitors are among the promising candidates that may help stabilize lysosomal homeostasis and alleviate metabolic and inflammatory stress in patients with HF [72,77]. Moreover, integrating lysosomal biomarkers into HF risk stratification models could provide valuable clinical insights, enabling early intervention and personalized treatment.
Further research should aim to clarify how lysosomal dysfunction contributes to distinct HF phenotypes, particularly HF with reduced ejection fraction (HFrEF) and HF with preserved ejection fraction (HFpEF). While lysosomal impairment in HFpEF may promote metabolic cardiomyopathy by disrupting autophagic clearance and increasing myocardial stiffness, excessive lysosomal membrane permeabilization in HFrEF may exacerbate inflammatory cascades and induce cardiomyocyte apoptosis. Understanding these differential mechanisms is essential for designing targeted interventions. Incorporating lysosomal-targeted therapies into standard HF treatment regimens requires a comprehensive evaluation of their safety, efficacy, and long-term impact on cardiac function. The interplay between lysosomal dysfunction, metabolic regulation, and myocardial remodeling underscores the need for translational studies to bridge preclinical discoveries with clinical applications. Future investigations should focus on determining optimal therapeutic strategies for modulating lysosomal function in HF, ultimately paving the way for novel treatment paradigms that address the underlying pathophysiology of HF at the lysosomal level.
- 10.5. Integrating Lysosomal Dysfunction into the Broader Landscape of CVD Pathophysiology
Lysosomal stress is not an isolated phenomenon but is closely linked to other pathophysiological processes, including ferroptosis, cellular senescence, and metabolic dysregulation [91]. Future studies should explore the interplay between lysosomal function and lipid metabolism, mitochondrial homeostasis, and immune activation to identify novel therapeutic targets [92]. Additionally, emerging evidence suggests that lysosomal dysfunction may play a role in the progression of age-related CVDs, such as atherosclerosis and diabetic cardiomyopathy [93]. Understanding how lysosomal integrity declines with age and how it contributes to CVD risk could provide new avenues for therapeutic intervention [94].
Targeting lysosomal dysfunction represents a promising frontier in cardiovascular medicine. Future research should prioritize the advancement of pharmacological interventions, the development of reliable biomarkers, the translation of lysosomal therapies into clinical practice, and the integration of lysosomal dysfunction into the broader landscape of CVD pathophysiology. By addressing these challenges, lysosome-targeted strategies have
the potential to redefine cardiovascular treatment paradigms and improve outcomes for patients with CVD.
Table 2. Future research avenues in lysosome-targeted cardiovascular therapies.
Category Key Focus Areas Advancing Lysosomal-Targeted Pharmacological Therapies
Clinical validation of TFEB activators, TRPML1 agonists, and NLRP3 inhibitors. Development of combination therapies with existing cardiovascular drugs. Optimization of dosing strategies to prevent maladaptive autophagy. Exploring Novel Biomarkers and Diagnostic Tools
Identification of circulating lysosomal biomarkers (cathepsins, LAMP2, TFEB activity). Development of advanced imaging techniques (PET tracers, MRI-based lysosomal assessment). Translating Strategies into Clinical Practice
Investigation of lysosomal dysfunction in heart failure subtypes (HFpEF vs. HFrEF). Evaluation of lysosomal-targeted therapies in randomized clinical trials. Integration of lysosomal biomarkers into cardiovascular risk stratification. Expanding the Role of Lysosomal Dysfunction in CVD Pathophysiology
Examination of the interplay between lysosomal stress, ferroptosis, and cellular senescence. Investigation of lysosomal impairment in age-related cardiovascular diseases (e.g., atherosclerosis, diabetic cardiomyopathy).
Author Contributions: T.O. conceptualized the review. All authors (T.O., K.-i.A. and T.T.) wrote the manuscript, and T.O. conducted the final revisions. All authors have read and agreed to the published version of the manuscript.
Funding: This study received no external funding. Acknowledgments: We would like to thank professional English language editors for their assistance in editing this manuscript. Conflicts of Interest: The authors declare no conflicts of interest.
Figures

Used In Evidence Reviews
Similar Papers
Molecules (Basel, Switzerland) · 2015
Curcumin, inflammation, and chronic diseases: how are they linked?
BioFactors (Oxford, England) · 2013
Curcumin in inflammatory diseases.
Drug discovery today · 2017
Curcumin and its topical formulations for wound healing applications.
Seminars in cancer biology · 2022
Curcumin and colorectal cancer: An update and current perspective on this natural medicine.
International journal of molecular sciences · 2018
Anti-Inflammatory Effects of Resveratrol: Mechanistic Insights.
Biomedicines · 2021